
Original Articles: 2021 Vol: 13 Issue: 1
Theoretical Bio-evaluation of 3, 5-dimethoxy-N-vinylbenzenamine Analogues as Potential anti- Sclerotinia sclerotiorum
Abel Kolawole Oyebamiji1, 6*, M Adelowo Joy1, Amao Folake Ayobami2, A Fadare Olatomide3, M Kaka Oluwatosin4, Oyawoye Olubukola Monisola4, A Oyedepo Temitope5, Semire Banjo6
1 Department of Basic Sciences, Adeleke University, P.M.B. 250, Ede, Osun State, Nigeria
2 Department of Mathematics, Faculty of Science, Adeleke University, P.M.B. 250, Ede, Osun State, Nigeria
3 Organic Chemistry Research Laboratory, Department of Chemistry, Obafemi Awolowo University, Ile-ife, Osun State, Nigeria
4 Department of Microbiology, Laboratory of Molecular of Biology, Immunology and Bioinformatics, Adeleke University, P.M.B. 250, Ede, Osun State, Nigeria
5 Department of Biochemistry, Adeleke University, P.M.B. 250, Ede, Osun State, Nigeria.
6 Computational Chemistry Laboratory, Department of Pure and Applied Chemistry, Ladoke Akintola, University of Technology, P.M.B. 4000,Ogbomoso, Oyo State, Nigeria
Abstract
Keywords
Capparis cartilaginea, Ulcerogenic; Indomethacin; Gastric Ulcer; Omeprazole; Rabbits
Introduction
The role of played by plants in the world cannot be overemphasized. It is one of the products produced by nature which human depended on so much for shelter, transportation, medicine and food stuffs etc. [1-3], series of plants like Barberry, Garlic, chamomile, ginger etc. are medicinal and they are very useful in treating several diseases like cancer, Diabetes mellitus, viral diseases, fungal disease, hypotension, Jaundice, malaria and fever [3-5].
Fungi are eukaryotic microbes which inhabit various locations like water, food and soil [6-7]. Several factors (moisture, temperature, pH, degree of aeration, quantity and kind of nutrients) which possess the ability to hinder the growth and distribution of fungi have been reported by many researchers [8]. Most of them are unobtrusive and this could be due to their lesser size as well as their ambiguous existences in soil and on lifeless substance [9].
Therefore, Sclerotinia sclerotiorum has been described by scientists as an imperative and disturbing fungus with wide environmental spreading [10]. As reported by Sharma [11], over 400 species of plants have been infested by Sclerotinia sclerotiorum and this has caused colossal damage to economy of many nations [11]. Also, several factors such as temperature and moisture levels etc. have been observed to influence the operation of Sclerotinia sclerotiorum in the soil. Therefore, understanding of its operation, distribution and its means of survival has assisted scientists in developing anti- Sclerotinia sclerotiorum agents [12].
Moreover, the universal use of quantum chemical method via density functional theory in investigating anti-fungal activities of drug-like molecules is still at its highest level [13] and this can be attribute it vastly meaningful part played in explaining the electronic structure and molecular compound’s reactivity [14]. Nevertheless, there are other methods in computational chemistry; however the proficient building of innovative measure for explaining and understanding chemical progressions has helped the frequent use of density functional theory method among scientists [15].
Therefore, this work is aimed at identifying molecular descriptors which describe antifungal activities of the studied compounds and to develop QSAR model for predicting biological activity observed compounds as well as observing molecular interaction between 3,5-dimethoxy-N-vinylbenzenamine analogues and Sclerotinia sclerotiorum .
Computational Details
Optimization
Equilibrium geometry of studied 3,5-dimethoxy-N-vinylbenzenamine derivatives were optimized using quantum chemical calculations via density functional theory with 6-31 G* as basis set. According to Becke [16], threeparameter density functional which comprise of Becke’s gradient exchange and the Lee, Yang, Parr correlation functional (i.e. B3LYP) [16] selection of basis set for quantum chemical calculation is a function of it precision [17]; thus, 6-31 G* basis set was used to optimized the studied compounds. The studied compounds are shown in Table 1.
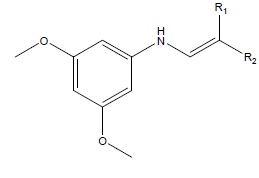 |
||
| R1 | R2 | |
| 1 | Ph | Ph |
| 2 | 4-tBu-Ph | 4-NO2-Ph |
| 3* | CH3 | 2-CH3-Ph |
| 4 | 3,4-2 Cl-Ph | 3-Py |
| 5 | 4-CH3-Ph | 4-Py |
| 6 | 3,4-2 CH3-Ph | 4-Py |
| 7* | 2-CH3-3-Cl-Ph | 4-Py |
| 8 | 4-Br-Ph | 4-Py |
| 9 | 4-Br-Ph | 2-Cl-3-Py |
| 10 | 4-CH3-Ph | 2-Cl-3-Py |
| 11* | 3,4-2 CH3-Ph | 2-Cl-3-Py |
| 12 | 4-CH3-Ph | 3-Cl-4-Py |
| 13 | 3,4-2 CH3-Ph | 3-Cl-4-Py |
| 14* | 4-Br-Ph | 3-Cl-4-Py |
| 15 | 4-Ph-Ph | 3-Cl-4-Py |
*denote test set
Table 1: Schematic structure of selected studied compounds
QSAR Studies
In this work, the obtained molecular descriptors from the optimized studied compounds were used to develop QSAR model (Table 2) so as to predict cytotoxicity of the observed compounds using partial least square methods via XLSTAT 2020.4.1.1020 (32 bit) [18] and Genetic Algorithm (GA) via MATLAB. The entire selected compounds were divided in two (Training set and test set) and series of arithmetic factors were considered for validation such as adjusted correlation coefficient, Mean Squared Error, p-value, F-value. Also, Absorption, Distribution, Metabolism, Excretion and the Toxicity properties of the studied 3,5-dimethoxy-N-vinylbenzenamine derivatives were done via online software (admetSAR)[19]. The factors considered were Blood Brain Barrier, Caco-2 cell permeability, Human Intestinal Absorption, Ames test. Also, six molecular compounds were proposed and their biological activities were predicted using the developed QSAR model.
| Equation | F | p-value | R2 | Adjusted R2 | MSE |
|---|---|---|---|---|---|
| IC50 = -624.32 - 6.24(DM) - 1.035(MW)+ 23.36(AREA) - 21.13(VOL) - 8.52(PSA) | 9.062 | p<0.0001 | 0.901 | 0.801 | 69.683 |
Table 2: Calculated QSAR model for the observed 3,5-dimethoxy-N-vinylbenzenamine derivatives
Molecular Docking Study
The molecular interaction between 3,5-dimethoxy-N-vinylbenzenamine derivatives and Sclerotinia sclerotiorum (PDB ID: 2 × 2 s) [20] were investigated using docking study. Series of software (EduPyMOL-v1.7.4.4-Win32, discovery studio 2017, autodock tool 1.5.6 and autodock vina 1.1.2) were used to accomplish this study. The observed grid box was as follows: center (X=43.197, Y=9.205, Z=8.724) and size (X=56, Y=40, Z=40) as well as the spacing was set to be 1.00 Å.
Results and Discussion
Calculated Molecular Descriptors
Series of molecular descriptors obtained from optimization of the studied compounds were reported. The calculated descriptors were highest occupied molecular orbitals energy (EHOMO) (eV), lowest unoccupied molecular orbital energy (ELUMO) (eV), dipole moment (Debye), molecular weight (amu), area (A2), volume (A3), polar surface area (PSA) (A2), Log P, polarizability, hydrogen bond donor, hydrogen bond acceptor (Table 3). The drug-likeness of the studied compounds was observed by considering Lipinski rule of five as reported by Adejoro [21]. The calculated Lipinski properties were Molecular Weight (≤ 500), Lipophilicity (Log P) (≤ 5), Hydrogen Bond Donor (HBD) (≤ 5), and Hydrogen Bond Acceptor (HBA) (≤ 10); thus, this showed that the studied compounds have drug-like properties.
Quantitative Structural Activity Relationship (QSAR) Study
The calculated molecular descriptors obtained from optimization of studied 3,5-dimethoxy-N-vinylbenzenamine were displayed in Table 3 and the correlation between the calculated descriptors were observed and investigated via Pearson correlation matrix. As shown in table 4, reasonable correlation was observed between the calculated molecular descriptors; therefore, dipole moment was fairly correlated with area, volume as well as polar surface area (PSA) by 0.780, 0.774 and 0.599 respectively. Also, Pearson’s correlation between molecular weight and area as well as volume is 0.641 and 0.613. As shown in Table 4, Area is fairly correlated to volume and polar surface area (PSA) by 0.996 and 0.726. More so, volume and polar surface area (PSA) are correlated to polar surface area and inhibition concentration (IC50) by 0.674 and -0.505 respectively.
| SNo. | EHOMO | ELUMO | DM | MW | AREA | VOL | PSA | LOG P | POL | HBD | HBA |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | -5.39 | -0.82 | 2.4 | 333.391 | 377.09 | 353.34 | 35.52 | 4.81 | 68.96 | 0 | 4 |
| 2 | -5.56 | -2.67 | 10.35 | 434.496 | 482.19 | 448.49 | 74.827 | 6.58 | 77.07 | 0 | 7 |
| 3 | -5.47 | -0.46 | 2.32 | 285.347 | 334.81 | 305.7 | 36.285 | 4.11 | 64.99 | 0 | 4 |
| 4 | -5.67 | -1.06 | 7.15 | 437.714 | 416.56 | 387.62 | 44.368 | 4.87 | 71.73 | 0 | 5 |
| 5 | -5.54 | -1.12 | 7.04 | 348.406 | 390.99 | 364.75 | 43.851 | 2.57 | 69.92 | 0 | 5 |
| 6 | -5.58 | -1.15 | 7.44 | 376.46 | 423.82 | 400.16 | 44.042 | 2.92 | 72.79 | 0 | 5 |
| 7 | -5.65 | -1.17 | 3.11 | 382.851 | 404.30 | 378.2 | 43.288 | 4.24 | 70.99 | 0 | 5 |
| 8 | -5.60 | -1.15 | 2.76 | 413.275 | 392.40 | 364.88 | 43.025 | 4.02 | 69.92 | 0 | 5 |
| 9 | -5.65 | -1.08 | 4.42 | 447.72 | 407.64 | 378.53 | 43.018 | 4.67 | 71.00 | 0 | 5 |
| 10 | -5.57 | -1.03 | 5.99 | 382.851 | 406.08 | 378.40 | 43.820 | 3.41 | 71.00 | 0 | 5 |
| 11 | -5.55 | -1.04 | 6.37 | 410.905 | 435.94 | 413.71 | 43.518 | 3.57 | 73.87 | 0 | 5 |
| 12 | -5.63 | -1.29 | 8.79 | 382.851 | 406.48 | 378.14 | 43.892 | 3.22 | 71.02 | 0 | 5 |
| 13 | -5.67 | -1.33 | 9.09 | 410.905 | 439.31 | 413.56 | 44.075 | 3.57 | 73.90 | 0 | 5 |
| 14 | -5.71 | -1.33 | 8.36 | 447.720 | 406.83 | 378.02 | 43.906 | 3.18 | 71.00 | 0 | 5 |
| 15 | -5.64 | -1.30 | 8.58 | 444.922 | 467.99 | 443.79 | 43.883 | 4.41 | 76.35 | 0 | 5 |
Table 3: Calculated descriptors for 3,5-dimethoxy-N-vinylbenzenamine derivatives
QSAR Model Analysis via Partial Least Square (PLS) and Genetic Algorithm (GA)
Series of molecular descriptors were obtained from the optimized molecular compounds and the selected descriptors which described anti- Sclerotinia sclerotiorum activities of 3,5-dimethoxy-N-vinylbenzenamine derivatives were used to develop QSAR model in order to predict the observed cytotoxicity. As shown in Table 5, the studied compounds were divided into two sets (test set and training set) in order to observe the efficiency of the developed model. The correlation between the predicted IC50 obtained using PLS and the observed IC50 showed the effectiveness of the developed QSAR model which was confirmed by the calculated correlation coefficient (R2) (0.901) since predictability of QSAR model could be confirmed by correlation coefficient (R2) ≤ 0.5 [22, 23]. Also, the catholic spread of the residuals on both sides of zero lines in the graph as displayed in figure 1 and figure 2 exposed that the developed QSAR model did not show any comparative imprecision. The predicted IC50 using partial least square were closer to the observed IC50, however, the predicted IC50 using genetic algorithm (GA) were much closer to the observed IC50 than the predicted IC50 using PLS which showed the efficiency of genetic algorithm in developing QSAR model and prediction (Figure 3). The selected molecular descriptors which described the anti- Sclerotinia sclerotiorum of the studied compounds proved to be effective as shown in the developed QSAR model as well as the contribution of each of the descriptor in the model were displayed in Figure 4. Also, the developed QSAR were validated by considering adjusted correlation coefficient (Adj R2), Mean square error (MSE), F-value and p-value.
| DM | MW | AREA | VOL | PSA | IC50 | |
|---|---|---|---|---|---|---|
| DM | 1.000 | |||||
| MW | 0.313 | 1.000 | ||||
| AREA | 0.780 | 0.641 | 1.000 | |||
| VOL | 0.774 | 0.613 | 0.996 | 1.000 | ||
| PSA | 0.599 | 0.401 | 0.726 | 0.674 | 1.000 | |
| IC50 | -0.203 | -0.140 | -0.320 | -0.311 | -0.505 | 1.000 |
Table 4: Pearson's correlation matrix for selected molecular descriptors
| Observed IC50 | PLR | GA | |
|---|---|---|---|
| 1 | 52.450 | 56.631 | 52.41845 |
| 2 | 11.340 | 12.141 | 11.3253 |
| 3* | 100.00 | 119.213 | 99.98545 |
| 4 | 33.220 | 41.326 | 33.20545 |
| 5 | 25.760 | 24.752 | 25.74719 |
| 6 | 10.970 | 10.370 | 10.95719 |
| 7* | 27.95 | 45.170 | 27.9441 |
| 8 | 25.760 | 21.517 | 25.7551 |
| 9 | 43.960 | 43.181 | 43.95712 |
| 10 | 60.940 | 60.027 | 60.93774 |
| 11* | 68.60 | -17.312 | 68.59793 |
| 12 | 65.890 | 56.781 | 65.88793 |
| 13 | 32.310 | 42.880 | 32.30836 |
| 14* | 53.20 | 2.87 | 53.19847 |
| 15 | 50.770 | 43.765 | 50.76847 |
| Carbendazim | 0.45 | - | - |
Table 5: Experimental and Calculated Inhibition concentration

Figure 1: Standardized residual against observed IC50

Figure 2: Standardized residual against predicted IC50

Figure 3: Calculated predicted IC50 against experimental IC50 via PLS

Figure 4: Graph of standardized coefficient against the variable.
Proposed Novel drug-like compounds
Series of drug-like compounds were proposed and these were achieved via addition of suitable derivatives to the studied parent compounds (Table 6). Also, IC50 was predicted for individual proposed compound using the developed QSAR model and the predicted IC50 were reported in Table 6. As shown in Table 6, the calculated inhibition efficiency for A2 and A3 proved to be more efficiency than the standard used.
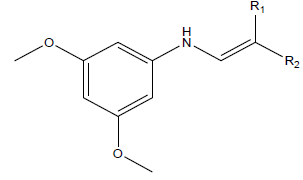 |
|||
| R1 | R2 | IC50 | |
| A1 | OCH3 | 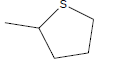 |
46.07 |
| A2 | OCH3 | 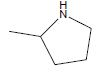 |
-9.79 |
| A3 | OCH3 | 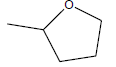 |
-6.10 |
| A4 | CH2F | C2H5 | 141.54 |
| A5 | CHF2 | C2H5 | 122.47 |
Table 6: Proposed drug-like compounds and predicted IC50
Molecular Docking Study
The molecular interaction between the studied compounds and Sclerotinia sclerotiorum (PDB ID: 2 x 2 s) were observed via molecular docking study. According to Ritchie [24] means of identifying drug-like molecule that has the capability to join with a receptor which is a function of calculated scoring describes molecular docking. Also, the degree of the connectivity between the studied 3,5-dimethoxy-N-vinylbenzenamine derivatives and Sclerotinia sclerotiorum (PDB ID: 2 × 2 s) could be enhanced by reducing scoring value [24]. Thus, it was observed that the derivatives attached to the parent compound used in this work were efficient and this could be confirmed via the calculated scoring values Figure 5 (Table 7). Also, comparing the binding affinity for the studied ligands to the standard used, it was observed that all the studied compounds have better ability to inhibit than Carbendazim (Standard). The binding affinity for compound 1-15 are -7.2 kcal/mol, -7.6 kcal/mol, -6.9 kcal/mol, -7.0 kcal/mol, -6.8 kcal/mol, -7.0 kcal/mol, -6.8 kcal/mol, -6.8 kcal/mol, -6.9 kcal/mol, -7.2 kcal/mol, -7.3 kcal/mol, -7.0 kcal/mol, -7.0 kcal/mol, -7.0 kcal/mol, -7.9 kcal/mol. The residues involved in the interaction between each ligand and the receptor as well as the type of non-bonding interaction was reported in Table 7.
| S NO | Scoring (kcal/mol) |
Residues involved in the interactions | Types of Non-bonding interaction involved |
|---|---|---|---|
| 1 | -7.2 | ALA-140, LYS-100, SER-117 | Conventional Hydrogen Bond, Carbon Hydrogen Bond, Pi-Sigma, Pi-Alkyl |
| 2 | -7.6 | LYS-100, ALA-140, SER-139 | Conventional Hydrogen Bond, Carbon Hydrogen Bond, Unfavourable Donor-Donor, Pi-Cation, Alkyl, Pi-Akyl |
| 3 | -6.9 | TRP-24, TYR-37, SER-44, THR-38 | Conventional Hydrogen Bond, Carbon Hydrogen Bond, Pi-Sigma, Pi-Pi- Stacked, Pi-Pi T-shaped, Pi-Alkyl |
| 4 | -7.0 | ALA-140, LYS-100, ASN-114 | Conventional Hydrogen Bond, Pi-Cation, Pi-Alkyl, Alkyl |
| 5 | -6.8 | ALA-140, LYS-100, SER-117 | Conventional Hydrogen Bond, Unfavorable Acceptor-Acceptor, Pi-Cation, Pi-Alkyl, Alkyl |
| 6 | -7.0 | ALA-140, GLN-121, LYS-100, ASN-113 | Conventional Hydrogen Bond, Carbon Hydrogen Bond, Pi-Cation, Pi-Donor Hydrogen Bond, Pi-Sigma, Pi-Alkyl |
| 7 | -6.8 | ALA—140, LYS-100, ASN-113, PRO-101 | Conventional Hydrogen Bond, Carbon Hydrogen Bond, Pi-Sigma, Pi-Alkyl |
| 8 | -6.8 | LYS-100, GLN-121, ALA-140 | Conventional Hydrogen Bond, Pi-Cation, Pi-Donor Hydrogen Bond, Pi-Sigma, Pi-Alkyl |
| 9 | -6.9 | LYS-100, ALA-140, GLY-120, PRO-101 | Conventional Hydrogen Bond, Carbon Hydrogen Bond, Pi-Cation, Pi-Alkyl, Alkyl |
| 10 | -7.2 | GLY-120, ALA-140, LYS-100 | Conventional Hydrogen Bond, Carbon Hydrogen Bond, Pi-Cation, Pi-Alkyl, Alkyl |
| 11 | -7.3 | GLY-120, ALA-140, LYS-100 | Conventional Hydrogen Bond, Carbon Hydrogen Bond, Pi-Cation, Alkyl, Pi-Alkyl |
| 12 | -7.0 | TYR-37, GLY-40, ALA-47, PRO-43, SER-44, TRP-24 | Conventional Hydrogen Bond, Pi-Pi stacked, Pi-Pi T-shaped, Alkyl, Pi-Alkyl |
| 13 | -7.0 | ALA-140, LYS-100 | Conventional Hydrogen Bond, Unfavorable Donor-Donor, Pi-Cation, Alkyl, Pi-Alkyl |
| 14 | -7.0 | ASN-46, TRP-24, TYR-37, ARG-35 | Conventional Hydrogen Bond, Carbon Hydrogen Bond, Pi-Pi Stacked, Pi-Pi T-shaped, Alkyl |
| 15 | -7.9 | LYS-100, ALA-140, GLN-121 | Carbon Hydrogen Bond, Pi-Cation, Pi-Alkyl |
| Carbendazim | -6.1 | VAL-6, LEU-53, GLY-58, TYR-107, TYR-63, ALA-61 | Conventional Hydrogen Bond, Pi-Pi T-shaped, Pi-Alkyl |
Table 7: Scoring and residues involved in the interaction between the studied complexes

Figure 5: Structure of the interactions between compounds 2, 11, 15 and Sclerotinia sclerotiorum (PDB ID: 2 x 2 s) respectively
Conclusion
Cytotoxity of fifteen molecular compounds were studied using quantum chemical method, QSAR and docking studies. The obtained descriptors described well the anti-Sclerotinia sclerotiorum activity of 3,5-dimethoxy-Nvinylbenzenamine derivatives and the obtained descriptors were further used to develop QSAR model. The developed model proved to be efficient via its predicting strength; however, genetic algorithm proved to be more predictive than PLS. Also, docking study reveals the molecular interaction between the studied ligands and the receptor; therefore, the result showed that all the studied compounds have better ability to inhibit Sclerotinia sclerotiorum than the standard.
Declarations
All authors contributed equally
Funding Statement
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Competing Interest Statement
The authors declare no conflict of interest.
References
- H Fallah-Hoseini; H Fakhrzadeh; B Larijani; A Shikhsamani. J Med Plant. 2006, 5, 1–8.
- RA Dar; M Shahnawaz; PH Qazi. The Journal of Phytopharmacol. 2017, 6(6), 349-351.
- M Rafieian-Kopaei. J HerbMed Plarmacol. 2012, 1(1), 1–2.
- AA Khosrokhawar; F Shamsa. Journal of Medicinal Plants. 2012, 9, 35.
- N karami; A Javid; BF Haghirosadat. Cancer press. 2017, 3 (1), 22-26
- NU Maheswari; R Komalavalli. J Curr Microbiol App Sci. 2013, 2, 135-141.
- FG Sartori; LF Leandro; LB Montanari. Curr Microbiol. 2013, 67, 107-111.
- G Gaddeyya; PS Niharika; P Bharathi; PKR Kumar. Adv Appl Sci Res. 2012, 3, 2020-2026.
- RT Moor. Botanica Marina. 1980, 23, 361–373.
- AS Rathi; M Jattan; R Punia; S Singh; P Kumar; R Avtar. Indian Phytopathol. 2018, 71, 407–413.
- P Sharma; PD Meena; PR Verma; GS Saharan; N Mehta; D Singh; A Kumar. J Oilseed Brassica. 2015, 6, 1–44.
- JP Angelique; AB Carl; IC Martin; KM Dean; SM Daren; AW Kiersten; DE Paul. J Integrated Pest Management. 2012, 3(2), 1-7.
- AK Oyebamiji; B Semire. Bull Pharm. Res. 2016, 6(3), 105-113.
- E Kraka; D Cremer. J Am Chem Soc. 2000, 122(34), 8245-8264.
- Kraka; D Cremer. J Am Chem Soc. 2000, 122(34), 8245-8264.
- D Jacquemin; E.A Perpete; I Ciofini; C Adamo. Acc Chem Res. 2009, 42(2), 326-334.
- AD Becke. J of phy Chem. 1993, 98, 5648–5652.
- KA Oyebamiji; B Semire. J New York Sci. 2016, 9(6), 58-66
- S Jie; C Feixiong; X You; L Weihua; TJ Yun, Chem. Inf Model. 2010, 50, 1034–1041
- G Sulzenbacher; V Roig-Zamboni; WJ Peumans; P Rougé; JM Els; V Damme; Y Bourne. J Mol Biol. 2010, 400, 715–723
- IA Adejoro; SO Waheed; OO Adeboye. J Phys Chem Biophys. 2016, 6, 213.
- AK Oyebamiji; OA Fadare; B Semire. Heliyon. Current Pharmaceutical Biotechnology. 2020, 6, 35-61.
- AK Oyebamiji; B Semire. Current Pharmaceutical Biotechnology. 2020, 21, 70-78.