
Perspective: 2024 Vol: 16 Issue: 10
Formulation and Charecterisation of Oral Dispersible Tablets of Fexofenadine Hydrochloride using Sublimation Technique
M. Sai Sri Vastav1*, Bhavya Reddi2, Renuka Devi Reddi3
Department of Pharmaceutical Sciences, Vignan Institute of Pharmaceutical Technology, Andhra Pradesh, India
- Corresponding Author:
- M. Sai Sri Vastav
Department of Pharmaceutical Sciences, Vignan Institute of Pharmaceutical Technology, Andhra Pradesh, India
Received: 23-Aug-2024, Manuscript No. JOCPR-24-146181; Editor assigned: 26-Aug-2024, PreQC No. JOCPR-24-146181 (PQ); Reviewed: 9-Sep-2024, QC No. JOCPR-24-146181; Revised: 16-Sep-2024, Manuscript No. JOCPR-24-146181 (R); Published: 23-Sep-2024, DOI:10.37532/0975-7384.2024.16(9).190
Citation: © 2024 Sai SVM, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Keywords
Fexofenadine hydrochloride; Oral dispersible tablets; Sublimation method; Direct compression method
Introduction
For many years, patients have been treated with pharmaceuticals in a variety of pharmaceutical dosage forms such as tablets, capsules, pills, suppositories, creams, ointments, liquids, aerosols, and injectable drug carriers. These traditional drug delivery mechanisms are still the most common pharmaceutical items found in the prescription and over-the-counter medicine markets.
It is well understood that this form of medication delivery method results in rapid drug release. To achieve and maintain drug concentrations within the therapeutically effective range required for therapy, this sort of drug delivery device is commonly used numerous times per day. As a result, there are substantial variations in medication levels.
Several technological advances have lately been made. They have fostered the development of innovative drug delivery techniques. These procedures have the capacity to control the rate of pharmaceutical administration, prolong the duration of a therapeutic impact, and/or direct drug delivery to a specific region. Despite the fact that these advancements have resulted in a number of novel drug delivery systems that have the potential to revolutionize the way medicine is administered and offer a number of therapeutic benefits, the terms "controlled release" and "sustained release" are occasionally used synonymously in technology. Unfortunately, in professional presentations and scientific journals over the years, these expressions have frequently been used synonymously.
Fast dissolving tablets
Oral administration is the most preferred mode of administration since it is simple to consume, painless, adaptable (suitable for a wide range of medication candidates), and, most importantly, patient-compliant. Furthermore, because they do not have to be made under sterile conditions, solid oral delivery devices are less expensive. Recent improvements in oral administration techniques have made it possible to address the physicochemical features of medications and improve patient compliance [1].
The formulation that is most easily grasped and swallowed by elderly patients is favoured. These requirements were considered when developing a quick dissolving tablet. It is easy to deliver such a tablet to any patient, regardless of age or location, because it can dissolve in just a small amount of water in the oral cavity. For instance, it can be carried by anyone who does not have ready access to water at any time and everywhere. It is also simple to dose elderly patients, patients who are confined to beds or newborns who have difficulty swallowing tablets and capsules [2]. With the capacity to accommodate a variety of physicochemical pharmacodynamic features of the pharmaceuticals, numerous firms have recently investigated and manufactured different types of fast dissolving tablets.
These tablets dissolve quickly in the mouth and are also known as Oro dispersible tablets, mouth dissolving tablets, fast dissolving tablets, rapid dissolving tablets, porous tablets, and rapid melts. Among the words described above, the United States Pharmacopeia (USP) designated these dosing forms as ODTS (Orally Disintegrating Tablets). The European Pharmacopeia has coined the term "Oro dispersible tablet" to designate tablets that dissolve quickly in the mouth and are ingested within 3 minutes. According to the FDA of the United States, MDT is a solid dosage form containing a medicine or active component that dissolves fast when placed on the tongue, sometimes within seconds. FDTS disintegration can take a few seconds or up to a minute.
Fast dissolving tablets characteristics
FDDTs are a unique dosage form that differs from more conventional dosage forms in a number of ways.
• The dosage form does not require water to be swallowed, which is quite practical, especially for patients. who are on the go and cannot easily acquire water.
• Compared to liquids, ease of administration and precise dose.
• Saliva in the mouth, throat, and esophagus can boost the bioavailability of certain medications.
• The pleasant tongue feels of these tablets, in especially for pediatric patients, helps to change the idea that medicine is typically thought of as a bitter pill.
• A higher bioavailability, especially in the case of hydrophobic and insoluble medicines.
• Rapid medication breakdown and absorption could result in rapid beginning of action.
• The ability to deliver liquid medication’s benefits in solid form [3].
Challenges of oral dissolving tablets
• The tablets lack sufficient mechanical strength. As a result, meticulous handling is essential throughout the manufacturing.
• If the tablets are not formed correctly, they could leave a bad taste or grittiness in the mouth.
• It is challenging to formulate drugs with larger doses into FDTs such as rifampicin (600mg), ethambutol (1000mg).
• Because the unit disintegrates quickly and the drug can be swallowed orally, it is unreasonable to expect drug delivery from a fast-dissolving formulation to prevent first-pass metabolism [4].
The requirement for the FDTs development
Patient factors: Patients (particularly children and the elderly) who are unable to swallow standard tablets and capsules with an 8-oz glass of water should take fast-dissolving dosage forms. They comprise of the following:
• Patient noncompliance because of choking fear.
• An eight-year-old allergy patient prefers a more effective antihistamine dosing form than a syrup.
• A middle-aged female undergoing radiation therapy for breast cancer may have nausea and be unable to take her H2-blocker.
• Older patients with depression may be unable to take solid doses.
• Experiencing chronic nausea while travelling or without access to water [4,5].
Effectiveness factor: Saliva-dissolving drugs are absorbed before reaching the stomach. Many medications are absorbed by the buccal, pharynx, and stomach. Any pre-gastric absorption inhibits first pass hepatic metabolism, increasing bioavailability. Furthermore, safety profiles may be improved for medications in which the oral cavity and pre-gastric segments of the GIT account for a significant portion of absorption, as well as medications in which significant amounts of hazardous metabolites are mediated by first-pass liver and stomach metabolism.
Materials and Methods
Fexofenadine hydrochloride was gifted from LEE pharma pvt ltd (Visakhapatnam, India). Cross povidone, Micro crystalline cellulose, Magnesium stearate was obtained from BMR chemicals, Hyderabad. Edible camphor was also obtained from BMR chemicals, Hyderabad. All other compounds, including solvents and reagents, are considered analytical grade.
Fourier transform infrared spectroscopy
To discover whether the medication and the polymers were compatible, FT-IR spectroscopy was used. Additionally, it was employed to look at the SPHs' chemical composition. The Fourier-Transform Infrared (FT-IR) spectrophotometer recorded FTIR spectra in the 400-4000 cm-1 range using the KBr pellet method. The FT-IR of the formulation and the pure medicine Fexofenadine hydrochloride were recorded. Each spectrum was created from a single average scan that was gathered between 400 and 4000 cm-1. Peak picking is allowed on the obtained spectrum [6-8].
Evaluation of powder blend
Pre-compression parameters: The combination was tested for attributes prior to being compacted into tablets.
Angle of repose: Solid flow characteristics have been described using the angle of repose. Angle of repose is one component of interparticle friction, which is the resistance to movement between particles. The surface of the powder or granule pile can only be tilted away from the horizontal plane at this maximum angle.
Tan ɵ = h/r
ɵ= Tan-1 h/r
Where, ɵ = angle of repose, h = height, r= radius.
A funnel is mounted over the platform at a height of 2-4 cm. The funnel's wall was progressively traversed by the loose powder until a powder cone formed. By measuring the height of the powder cone and the radius of the powder heap, the angle of repose was calculated [9].
Bulk density: By carefully pouring a sample of the medication via a glass funnel into a 50ml graduated cylinder, it was possible to calculate the bulk density of the substance. The samples' total volume was recorded.

Tapped density: The tapped density was calculated with an Electro Lab density tester, which is made up of a granular cylinder fixed on a mechanical tapping mechanism. A correctly weighed sample of powder was transferred to the cylinder using a funnel. Typically, after recording the initial volume, the sample is tapped (50, 750, or 1250 times) until no additional volume loss is seen or the percentage fluctuation is less than 2%.
For the substance in question, there should be enough taps used to ensure reproducibility. The volume was recorded, and the formula for calculating tapped density was used:
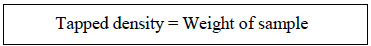
Compressibility index and hausner’s ratio: The compressibility index and its associated Hausner's ratio have recently emerged as the most simple, rapid, and popular methods for projecting powder flow characteristics. Because all of these variables can influence the measured compressibility index, it has been proposed as an indirect measure of bulk density, size and shape, surface area, moisture content, and material cohesiveness. The compressibility index and Hausner's ratio could be calculated from the powder's tapped density and bulk density.


Evaluation of post compression parameters
The formulation was assessed for weight variation, hardness, friability, drug content estimation, in vitro dispersion time, wetting time, water absorption ratio, tablet disintegration time, and in vitro drug release.
| Ingredients (%w/w) | F1 | F2 | F3 | F4 | F5 | F6 | F7 | F8 | F9 |
|---|---|---|---|---|---|---|---|---|---|
| Fexofenadine HCL | 60 | 60 | 60 | 60 | 60 | 60 | 60 | 60 | 60 |
| Camphor | 15 | - | 7.5 | 22.5 | - | 11.25 | 30 | - | 15 |
| Cross povidone | - | 15 | 7.5 | - | 22.5 | 11.25 | - | 30 | 15 |
| MCC | 66 | 66 | 66 | 63.5 | 63.5 | 63.5 | 56 | 56 | 56 |
| Mg Stearate | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| Total weight | 150 | 150 | 150 | 150 | 150 | 150 | 150 | 150 | 150 |
Table 1: Formulation of Fast dissolving tablets of Fexofenadine HCL.
Weight variation: Ten tablets were picked at random and each weighed individually. The typical weight of ten tablets was determined. The average tablet weight was compared to the individual tablet weights.

Hardness: The diametric compression test determines the amount of force necessary to fracture a tablet in a given situation. A tablet was placed between two anvils of a hardness tester, and the amount of force required to break it was measured in N (kg/cm2). To calculate it, six tablets of each formulation are consumed. Monsanto hardness tester, which applies force to the tablet in two opposed directions via an internal spring.
Friability: To endure the mechanical shock of handling during production, packaging, and shipping, tablets must have a particular level of strength or hardness and resistance. The formula was used to compute the percentage of friability after a pre-weighed sample of five tablets was placed in a friabilator and operated for one hundred rotations [10-12]:
Where W0 – Weight of tablet before test W – Weight of tablet after test
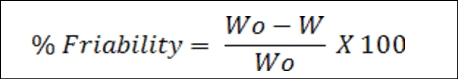
Estimation of drug content: To ensure dosage unit homogeneity, each unit in a batch should have active ingredient content that falls within a restricted range of label claims. Dosage forms that include a single dose or a portion of a dose of an active component are referred to as dosage units. The degree of consistency for medications across dosage units is referred to as "uniformity of drug content." A 10mg-equivalent oral dispersible tablet of fexofenadine HCL was weighed and transferred to a 10ml volumetric flask. The solution was filtered and diluted before being tested for drug content using a UV-Visible spectrophotometer at 212nm.
In vitro dispersion time: The in vitro dispersion time was calculated by inserting a tablet in a petri dish filled with 10ml of 6.8 PH phosphate buffer. Three tablets from each formulation were randomly selected and tested. The duration of in vitro dispersion is measured and expressed in seconds [13].
Wetting time and water absorption ratio: The wetting time of the dosage form is linked to the contact angle. Wetting time is another important factor to consider when determining the disintegration properties of mouth-dissolving tablets; a shorter wetting time indicates that the tablet will disintegrate faster. There is a simple way for determining the pill's wetting time. The two circular tissue papers are placed within a petri dish with the same inner diameter. The petri dish was treated with a 6.8 pH phosphate buffer containing ten millilitres of water-soluble dye. A pill was carefully placed on the tissue's surface to keep it from being submerged in the solution. The length of time it took for the solution to attain saturation is then recorded as the soaking time [14].
Tablet disintegration time: The time it takes for the tablet to dissolve is one of the most important requirements for the fast-dissolving tablets. These dosage forms need to dissolve in under a minute. The disintegration time was calculated using disintegration test equipment. The apparatus consisted of six tubes, with a tablet and a disc attached to each tube. The time in seconds that the pill has to totally disintegrate such that there isn't any visible mass inside the gadget [15].
In Vitro drug release studies: The in vitro dissolution study followed the guidelines of the United States Pharmacopoeia (USP). The paddle method was used to investigate medication release from tablets. The dissolving medium contained 900ml of 6.8PH phosphate buffer. The discharge was carried out at 37°C ± 0.5°C and 50 RPM.5ml samples were removed at predefined time intervals (0 to 1.5h), replaced with fresh medium, and analysed for Fexofenadine HCL using a UV spectrophotometer at 212nm. The percent drug release was estimated using the medication's calibration curve in 6.8PH phosphate buffer [16-18].
Evaluation of tablet
| Formulations | Angle of repose | Bulk density (gm/ml) | Tapped density (gm/ml) | Carr’s Index | Hausner’s ratio |
|---|---|---|---|---|---|
| F1 | 23.32±1.10 | 0.284±0.03 | 0.323±0.05 | 12.03±0.10 | 1.13±0.01 |
| F2 | 24.18±1.34 | 0.289±0.10 | 0.312±0.06 | 11.15±0.55 | 1.10±0.02 |
| F3 | 21.16±1.26 | 0.283±0.02 | 0.325±0.12 | 12.96±1.8 | 1.15±0.01 |
| F4 | 21.01±0.46 | 0.280±0.54 | 0.317±0.10 | 14.05±0.56 | 1.13±0.01 |
| F5 | 19.35±1.17 | 0.293±1.10 | 0.331±0.72 | 11.48±0.51 | 1.31±0.01 |
| F6 | 25.35±0.49 | 0.284±0.76 | 0.322±0.38 | 13.43±0.52 | 1.13±0.02 |
| F7 | 21.06±1.12 | 0.285±0.75 | 0.324±1.13 | 14.46±0.61 | 1.15±0.02 |
| F8 | 23.12±1.29 | 0.280±0.25 | 0.320±1.05 | 11.98±0.54 | 1.14±0.01 |
| F9 | 24.32±1.32 | 0.289±0.3 | 0.321±0.58 | 12.03±0.71 | 1.11±0.01 |
Table 2: Evaluation of pre-compression parameters.
| Formulae | Weight variation test (%) | Friability (%) | Hardness kg/cm2 | Thickness(mm) |
|---|---|---|---|---|
| F1 | 147±0.20 | 0.46 | 3.79±0.15 | 3 |
| F2 | 145±0.89 | 0.47 | 3.62±0.26 | 3 |
| F3 | 149±0.75 | 0.33 | 3.53±0.14 | 3 |
| F4 | 146±0.23 | 0.6 | 3.96±0.25 | 3 |
| F5 | 148±0.2 | 0.53 | 3.76±0.22 | 3 |
| F6 | 145±0.25 | 0.4 | 3.84±0.26 | 3 |
| F7 | 146±0.20 | 0.67 | 3.36±0.34 | 3 |
| F8 | 148±0.75 | 0.4 | 3.71±0.25 | 3 |
| F9 | 149±0.23 | 0.66 | 3.43±0.20 | 3 |
Table 3: Post compression parameters of all ODT tablets.
| Formulae | Disintegration time | In vitro dispersion time | Drug content (%) |
|---|---|---|---|
| F1 | 9 | 50 | 98.7 |
| F2 | 8 | 32 | 99.1 |
| F3 | 8 | 45 | 98 |
| F4 | 9 | 55 | 99.15 |
| F5 | 7 | 38 | 98.32 |
| F6 | 6 | 35 | 98.57 |
| F7 | 6 | 48 | 98.2 |
| F8 | 5 | 47 | 98.4 |
| F9 | 5 | 30 | 99.8 |
Table 4: Post compression parameters of all ODT tablets.
In Vitro release kinetics:
| Time | F1 | F2 | F3 | F4 | F5 | F6 | F7 | F8 | F9 |
|---|---|---|---|---|---|---|---|---|---|
| (min) | |||||||||
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 5 | 9.91 | 11.81 | 13.85 | 12.09 | 14.11 | 16.2 | 15.36 | 17.24 | 19.8 |
| 10 | 20.32 | 22.04 | 24.35 | 23.59 | 25.14 | 27.82 | 26.53 | 28.64 | 30.35 |
| 15 | 27.28 | 29.35 | 31.26 | 30.17 | 32.31 | 34.67 | 34.35 | 36.53 | 38.2 |
| 30 | 54.67 | 56.47 | 58.41 | 57.28 | 59.48 | 61.34 | 60.51 | 62.19 | 65.4 |
| 45 | 71.38 | 73.19 | 75.48 | 73.33 | 76.57 | 78.47 | 76.19 | 78.29 | 80.1 |
| 60 | 80.77 | 82.78 | 84.75 | 82.97 | 85.17 | 87.25 | 86.46 | 89.91 | 99.45 |
Table 5: In Vitro drug release studies of all ODT formulations.
All nine formulations of Fexofenadine HCL tablets were subjected to in vitro release tests, which were carried out using dissolution media. Use the USP-2 (paddle type) dissolve device to prepare a pH 6.8 phosphate buffer solution. The results were analyzed for 60 minutes. The F9 formulation has the highest release at 60 minutes, with 99.45%. All other metrics, such as hardness, thickness, friability, drug content, and weight fluctuation, were within the range. As a result, formulation F9 was chosen as the optimal formulation. The data for all of the formulated tablets is presented in the tables below, and graphs are produced accordingly.
Zero Order:

Figure 1: In vitro drug release of F1 to F3.

Figure 2: In vitro drug release of F4 to F6.

Figure 3: In vitro drug release of F7 to F9.
First order release kinetics:
| Time | F1 | F2 | F3 | F4 | F5 | F6 | F7 | F8 | F9 |
|---|---|---|---|---|---|---|---|---|---|
| (min) | |||||||||
| 0 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 5 | 1.954 | 1.945 | 1.935 | 1.944 | 1.933 | 1.923 | 1.927 | 1.853 | 1.904 |
| 10 | 1.901 | 1.891 | 1.878 | 1.883 | 1.874 | 1.858 | 1.866 | 1.802 | 1.842 |
| 15 | 1.861 | 1.849 | 1.837 | 1.844 | 1.83 | 1.815 | 1.817 | 1.577 | 1.79 |
| 30 | 1.656 | 1.638 | 1.618 | 1.63 | 1.607 | 1.587 | 1.596 | 1.336 | 1.539 |
| 45 | 1.456 | 1.428 | 1.389 | 1.426 | 1.369 | 1.333 | 1.376 | 1.003 | 1.298 |
| 60 | 1.283 | 1.236 | 1.183 | 1.231 | 1.711 | 1.105 | 1.131 | 1.195 | 0.877 |
Table 6: First order drug release kinetics studies of all ODT formulations.

Figure 4: First order drug release of F1 to F3.

Figure 5: First order drug release of F4 to F6.

Figure 6: First order drug release of F7 to F9.
Higuchi data:
| Time | F1 | F2 | F3 | F4 | F5 | F6 | F7 | F8 | F9 |
|---|---|---|---|---|---|---|---|---|---|
| (min) | |||||||||
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2.23607 | 9.91 | 11.81 | 13.85 | 12.09 | 14.11 | 16.2 | 15.36 | 17.24 | 19.8 |
| 3.16228 | 20.32 | 22.04 | 24.35 | 23.59 | 25.14 | 27.82 | 26.53 | 28.64 | 30.35 |
| 3.87298 | 27.28 | 29.35 | 31.26 | 30.17 | 32.31 | 34.67 | 34.35 | 36.53 | 38.2 |
| 5.47723 | 54.67 | 56.47 | 58.41 | 57.28 | 59.48 | 61.34 | 60.51 | 62.19 | 65.4 |
| 6.7082 | 71.38 | 73.19 | 75.48 | 73.33 | 76.57 | 78.47 | 76.19 | 78.29 | 80.1 |
| 7.74597 | 80.77 | 82.78 | 84.75 | 82.97 | 85.17 | 87.25 | 86.46 | 89.91 | 99.45 |
Table 7: Higuchi data of all ODT formulations.

Figure 7: Higuchi plot of F1 to F3.

Figure 8: Higuchi plot of F4 to F6.

Figure 9: Higuchi plot of F7 to F9.
Korsmeyer-peppas data:
| Time | F1 | F2 | F3 | F4 | F5 | F6 | F7 | F8 | F9 |
|---|---|---|---|---|---|---|---|---|---|
| (min) | |||||||||
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 0.69897 | 0.996 | 1.072 | 1.141 | 1.082 | 1.149 | 1.209 | 1.186 | 1.236 | 1.296 |
| 1 | 1.307 | 1.343 | 1.386 | 1.372 | 1.4 | 1.444 | 1.423 | 1.456 | 1.482 |
| 1.176091 | 1.435 | 1.467 | 1.494 | 1.479 | 1.509 | 1.539 | 1.535 | 1.562 | 1.582 |
| 1.477121 | 1.737 | 1.751 | 1.766 | 1.758 | 1.774 | 1.787 | 1.781 | 1.793 | 1.815 |
| 1.653213 | 1.853 | 1.864 | 1.877 | 1.865 | 1.884 | 1.894 | 1.881 | 1.893 | 1.903 |
| 1.778151 | 1.907 | 1.917 | 1.928 | 1.918 | 1.93 | 1.94 | 1.936 | 1.953 | 1.965 |
Table 8: Peppas data of all ODT formulations.

Figure 10: Peppas plot of F1 to F3.

Figure 11: Peppas plot of F4 to F6.
Results and Disscussion
The in vitro dissolution data for the best formulation F9 were fitted in various kinetic models, including zero order, first order, Higuchi, and Korsmeyer-Peppas equations. The optimized formulation F9 has an R2 value of 0.952, which is closer to the '1' that is conformed since it follows the Zero order release.

Figure 12: Peppas plot of F7 to F9.
| R2 Values | n Values | ||||
|---|---|---|---|---|---|
| Formulation | Zero order | First order | Higuchi | Korsmeyer- Peppas | Korsmeyer- Peppas(n) |
| F9 | 0.952 | 0.982 | 0.987 | 0.916 | 1.047 |
Table 9: R2 Values of optimised formulation.
The Higuchi and Peppas figure, which defines Case I or Fickian diffusion as 0.45 n, anomalous behavior or Non-Fickian transport as 0.45 n 0.89, Case II transport as 0.89 n, and Super Case II transport as 0.89 n, provides additional evidence for the drug release mechanism. The optimized formulation (F9) has a value of 1.047, or "n value is >0.89," indicating non-Fickian transport. The accompanying table shows the release kinetics for the enhanced formulation.
Pre-formulation studies
Melting point determination: The capillary method was used to determine the melting point of Fexofenadine HCl. The melting point of fexofenadine HCl was discovered to be around 190 °C, meeting BP requirements and indicating that the medication sample was pure.
Standard curve of fexofenadine HCL: The ƛmax was found at 212 nm. Thus, at this wavelength, the Fexofenadine HCL standard curve was developed. The standard curve was linear in the concentration range of 0 to 25 g/ml. The standard curve in 6.8PH phosphate buffer was constructed by plotting absorbance against concentration at 212 nm and is consistent with Beer's law. R2 was found to be 0.952.
Angle of repose: The precompression blend's angle of repose was computed, and the results are shown in Table 13. All formulation mixes were discovered to fall between the values of 23.50 and 33.60, indicating that blends have adequate flow characteristics.
Carr’s index: A calculation of Carr's index revealed a range of 5.6%-23.84%. The findings in Table No. 13 show that the powder mixes have the necessary flow characteristics for compression.
Hausner’s ratio: The calculation of Hausner's Ratio revealed a range of 1.07 to 1.31. The outcomes displayed in Table No. 13 revealed that the powder mixes' superior flow characteristics for compression.
Post compression studies
Evaluation of tablets:
• Shape of the tablets: Tablets in all formulations were found to be spherical, without apparent cracks, and white in colour.
• Thickness of tablet: According to table no. 14, all formulations' thickness was determined to range from 3 to 3.5 mm.
• Hardness test: Tablet hardness was assessed in each batch for formulations F1 through F9 and ranged from 3.1 to 4.2 kg/cm2. The pills' hardness varied from 3.1 to 4.2 kg/cm2. This guarantees all batches will have good handling characteristics.
• Friability: Table No. 14 is a summary of the results of the friability test. It lies between 0.46% and 0.75%. All of the formulations' friability levels were less than 1%, confirming the mechanical stability of the tablets.
• Weight variation test: In table no. 14, the % weight variation for each formulation was tallied. As long as the percentage of weight fluctuation stayed within the pharmacopeial guidelines of 7.5% of the average weight, all of the manufactured pills passed the weight variation test. All of the tablets had consistent weights with small standard deviation values.
• Drug content uniformity: The drug content percentages for F1 through F9 ranged from 98 to 99.2%. It adheres to the IP limitations.
• Disintegration test: The results of the Fexofenadine HCl tablet disintegration test was recorded in table 15. All of the tablets' in vitro disintegration time measurements were found to be within the permitted ranges and meet the requirements for orally dispersible tablets. The formulation exhibits a shorter disintegration time at the highest crospovidone and camphor concentrations, it was found. Cross povidone speeds up disintegration by facilitating the wicking effect of camphor.
• Wetting time: The wetting time for all the formulations were mentioned in table 15. The value lies in the range of 42-64.
• In Vitro dissolution study: Using a dissolution equipment in a 6.8ph phosphate buffer, in vitro release tests of all the fast-dissolving tablet formulations of fexofenadine HCl were performed. All of the formulations' obtained release data were summarized in a table. displays the cumulative percent of the drug released as a function of time for various formulations. The formulation F9, which contains 20% crospovidone and 20% camphor, has a greater drug release at the end of 60 minutes (92.14%). According to the study's findings, an increase in the concentration of super disintegrants and sublimating agents resulted in a rise in the overall percentage of drugs dissolved.
• Stability study: According to ICH requirements, stability assessments of the formulations were conducted. Stability tests on the improved formulation F9 were conducted for a month at 40°C and 75% RH. The appearance and change in drug content were used to evaluate the chemical stability and physical stability, respectively. The 19 results displayed showed good stability. F9 was thought to be the best formula based on the results mentioned above.
Conclusion
The present investigation can lead to the following conclusions
• Camphor was utilized as a sublimating agent in the formulation of Fexofenadine HCl fast-dissolving tablets, and super disintegrating agents were shown to be efficient in reducing chipping, capping, and sticking.
• When 20% camphor and 20% cross povidone was used, it showed higher dissolution rate compared to other formulations (10%, 15%).
• When the super disintegrants concentrations were increased from 10% to 20%, increase in dissolution rate was observed.
• When compared to different super disintegrants the dissolution rate was decreasing in the following order- Mixture > Cross povidone > Camphor.
• Formulation F9 demonstrated fairly good release, hardness, low friability, and the shortest wetting and disintegration times when compared to other formulations. Hence, F9 is regarded as the finest formulation.
• Infrared spectroscopy investigations showed that the medication is compatible with the other formulations.
• It was decided that Fexofenadine HCl oral dispersible tablets could be manufactured effectively since they fit all of the characteristics for an oral dispersible tablet and would be an alternative to the conventional tablets that are currently available.
• The produced formulations were stable over a one-month storage period at 40°C and 75% humidity.
• After dissolution studies, formulation F9 drug release is very close to the drug release of the marketed formulation. So, F9 is considered as the optimized formulation.
• So, it was determined that the combination of super disintegrants and the sublimation procedure were ideal methods for creating fast-acting tablets of Fexofenadine HCl which is an Anti-Histamine drug.
References
- Habib W, Khankari R, Hontz J. Crit Rev Ther Drug Carrier Syst. 2000;17(1).
[Crossref] [Google Scholar] [PubMed]
- Seager H. J Pharm Pharmacol. 1998;50(4):375-382.
[Crossref] [Google Scholar] [PubMed]
- L V Allen. Int J Pharm Compd. 1997;(1):90-92.
[PubMed]
- Chang RK, Guo X, Burnside BA, et al. Pharm Technol. 2000;24(6):52-58.
- Slowson M, Slowson M. Pharm Times. 1985:90-96.
- Karpe M, Mali N, Kadam V. J Appl Pharm Sci. 2012:101-105.
- Shukla D, Chakraborty S, Singh S, et al. Scientia Pharmaceutica. 2009;77(2):309-326.
- Drug OD. Center Drug Eval Res. 2011;1000.
- Furtado S, Deveswaran R, Bharath S, et al. Trop. j. pharm. res. 2008;7(4):1185-1189.
- Pawar PB, Mansuk AG, Ramteke KH, et al. Int J Herb Med. 2011;1(2):22-29.
- Habib W, Khankari R, Hontz J. Crit Rev Ther Drug Carrier Syst. 2000;17(1)
- Sreenivas SA, Gadad AP, Dandagi PM, et al. Indian Drug. 2006;43(1):35.
- Bhatti A. Indian Drug. 2006;43(8):643.
- Kumar R, Patil M B, Sachin R, et al. J en Pract. 2015;3(208):2.
- Venkatesh DP. Asian J. Pharm.
- Jha SK, Vijayalakshmi P, Karki R, et al. Asian J. Pharm. 2008;2(4).
- Malke S, Shidhaye S, Kadam VJ. Indian J Pharm Sci. 2007;69(2).
- Chaudhari SP, Chaudhari PD, Kolsure PK. Indian Drug. 2006;43(12):966.