
Original Articles: 2023 Vol: 15 Issue: 5
Liquid Chromatography Tandem Mass Spectrometry Method for Quantitation of Ivermectin and Simultaneous Detection of Albendazole in Human Plasma
Adam M Fimbo1,2, Eulambius M Mlugu3, Rajabu Hussein Mnkugwe4, Gerald S Kulwa2, Mohamed A Iwodyah2, Peter P Kunambi4, Alpha Malishee5, Appolinary AR Kamuhabwa6, Eleni Aklillu1, Omary M Minzi6*
1Department of Laboratory Medicine, Karolinska Institute, Huddinge, Stockholm, Sweden
2Department of Medical Science, Muhimbili University of Health and Allied Sciences, Dar es Salaam, Tanzania
3Department of Pharmaceutics and Pharmacy Practice, Muhimbili University of Health and Allied Sciences, Dar es Salaam, Tanzania
4Department of Clinical Pharmacology, Muhimbili University of Health and Allied Sciences, Dar es Salaam, Tanzania
5Department of Clinical Pharmacology, National Institute for Medical Research, Tanga, Tanzania
6Department of Clinical Pharmacy and Pharmacology, Muhimbili University of Health and Allied Sciences, Dar es Salaam, Tanzania
- Corresponding Author:
- Omary M Minzi
Department of Clinical Pharmacy and Pharmacology
Muhimbili University of Health and Allied Sciences
Dar es Salaam, Tanzania
Received: 04-Feb-2023, Manuscript No. JOCPR-23-88653; Editor assigned: 06-Feb-2023, PreQC No. JOCPR-23-88653 (PQ); Reviewed: 20-Feb-2023, QC No. JOCPR-23-88653; Revised: 26-Jun-2023, Manuscript No. JOCPR-23-88653 (R); Published: 03-Jul-2023
Citation: Fimbo AM, et al. 2023. Liquid Chromatography Tandem Mass Spectrometry Method for Quantitation of Ivermectin and Simultaneous Detection of Albendazole in Human Plasma. J. Chem Pharm. Res., 15:036.
Copyright: © 2023 Fimbo AM, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Ivermectin (IVM) is an antiparasitic drug administered with Albendazole (ALB) in the public health programme for the control of Lymphatic Filariasis (LF) in sub-Saharan Africa. The effectiveness of IVM in the programme depends in part on its Pharmacokinetic (PK) disposition which needs a sensitive bioanalytical method to analyses the same in the presence of ALB. A sensitive LC-MS/MS method was successful developed and quantified IVM accurately (bias<14) and precisely (CV<8) using matrix matched calibration curve at a concentration range of 0.5 ng/mL-200 ng/mL with correlation coefficient (r2) of 0.999. ALB, abamectin (internal standard) and IVM were eluted at 1.0, 4.3 and 6.2 min, respectively with a total run time of 8 min. The method was validated according to the FDA guidelines on bioanalytical method validation. A 2.1 mm × 100 mm, 1.8 µm id Agilent Zorbax RRHD Eclipse plus C18 column in gradient elution mode at a flow rate of 0.3 mL/min with a mobile phase containing acidified ammonium formate buffer (0.1% formic acid) and acetonitrile was used. The uniqueness of this method is a good resolution achieved through precipitation of proteins and extraction of analytes at lower temperature and also its ability to successfully obtain pharmacokinetics data from individuals who took IVM in the field after Mass Drug Administration (MDA) in north-eastern part of Tanzania. The method has been successfully used by our research group to assess pharmacokinetics of individuals that were exposed under ivermectin mass drug administration in Tanzania.
Keywords
Ivermectin, Albendazole, LC-MS/MS, Human plasma, Pharmacokinetic (PK)
Introduction
Ivermectin (IVM), chemically known as 22, 23-dihydro-avermectin is a mixture of two semisynthetic analogs resulting from fermentation of the bacteria Streptomycetes avermitilis [1]. It is an antiparasitic drug deployed during Mass Drug Administration (MDA) campaigns for the control of Lymphatic Filariasis (LF) in sub-Saharan Africa. During MDA, IVM is co-administered with Albendazole (ALB) a benzimidazole broad spectrum anthelmintic for treatment of soil transmitted helminthes. Both IVM and ALB are included in the WHO essential medicines list and approved for the treatment and control of parasitic infections in humans including LF [2,3].
For successful pharmacokinetic-pharmacodynamics studies, a well-established sensitive and selective method to quantify IVM in the presence of ALB is needed. Several methods have been developed for quantification of IVM in various matrices. High Performance Liquid Chromatography with Fluorescence (HPLC-FL) and Gas Chromatography Mass Spectrometry (GC-MS) for quantification of IVM are reported. However, these methods have some limitations as they require derivatization which is laborious, time consuming and expensive to run in resource constrained laboratories. A number of Liquid Chromatography tandem Mass Spectrometry (LC-MS/MS) methods for quantification of IVM have been described including in aquatic environment, environmental water, fish tissue, dogs, milk, bovine tissue monkey and mouse plasma as well as in humans [4-11]. A solid phase extraction method was reported by Chhonker, et al in 2018 [12]. However, bioanalytical methods that employ solid phase extraction procedure are time consuming and expensive to run in the laboratories of resource limited countries.
LC-MS/MS methods for quantification of IVM that apply liquid-liquid extraction procedure in dog plasma, human plasma and milk are available in literature [13,14]. Nevertheless, when we tried to set the methods in our laboratory we experienced mounting challenges including failure to reproduce the extraction procedure so as to achieve good recovery and accuracy despite introducing various modifications. We therefore optimized the conditions and managed to develop a method that worked well in our laboratory.
The method we here describe allows for quantification of IVM without interference of ALB in human plasma samples. The method exhibited acceptable accuracy with minimum bias. The applicability of the method was tested using human plasma samples collected from the field amongst individuals exposed to MDA drugs in north-eastern part of Tanzania. The method can be used to monitor PK-PD of IVM in individuals.
Materials and Methods
Chemicals and Reagents
IVM was purchased from the European directorate for the quality of medicines (Kastner, Strasbourg, France). Abamectin as an Internal Standard (IS) was purchased from Dr. Enhrenstorfer GmbH chemicals company (Augsburg, Germany) (Figure 1). ALB was purchased from the United States pharmacopeia (Rockville, Maryland, United States) while acetonitrile (LC-MS grade) and formic acid from chem lab (de Arend, Zedelgem, Belgium). Ammonium formate was purchased from sigma aldrich (St. Louis, Missouri, United States) and n-hexane from fisher chemical (Eastbourne, UK). Purified water (0.068 µS/cm) was freshly prepared in the laboratory using evoqua ultrapure water purification system, reverse osmosis (Gunzburg, Deutschland, Germany). Blank human plasma from six different individuals was donated by Bugando medical centre blood bank, Mwanza, Tanzania (Figure 1).

Figure 1: The chemical structures of (A) Ivermectin component B1a and B1b; (B) Abamectin and; (C) Albendazole.
Instrumentation
Agilent LC system 1290 infinity II equipped with high speed pump (G7120A), column oven MTC (G7116B) along with a vial autosampler (G7129B) was used. Mass spectrometric detection was performed on an agilent MS 6460 system (Santa Clara, California, United States) equipped with jet stream source in positive electrospray ionization mode (ESI+) with capillary voltage set at 5 kV, drying gas (N2) 5 L/min, drying gas temperature of 250°C, nebulizer 30 psi, sheath gas heater 200°C and sheath gas flow at the rate of 7 L/min. The MS/MS system was operated at a unit resolution in the Multiple Reaction Monitoring (MRM) mode. All analytes were detected in the positive ionization mode utilizing ESI+ with the indicated conditions above.
The MRM precursor ion→product ion transitions for IVM, ALB and IS in m/z and their corresponding retention time and Collision Energy (CE) are presented in Table 1. Quantitation was performed using mass hunter workstation data acquisition software B.08.02 (Santa Clara, California, United States).
| Analyte | Retention time | Precursor ion (Q1) m/z | Product ion (Q3) m/z | Collision energy (eV) |
|---|---|---|---|---|
| Albendazole | 1.086 | 266 | 233.9 | 15 |
| 190.8 | 35 | |||
| Abamectin | 4.365 | 890.5 | 566.7 | 10 |
| 305 | 20 | |||
| Ivermectin | 6.227 | 892.5 | 568.9 | 10 |
| 306.9 | 20 |
Table 1: Summary of MS parameters: Precursor ion, product ion, retention time and collision energy.
Chromatographic conditions
Several solvents including methanol, acetonitrile and ammonium formate were tested to find suitable mobile phases with increased sensitivity, improved peak resolution and shorter run times. Eventually, optimum separation was achieved using a mobile phase consisting of solvent A (2 mM ammonium formate with 0.1% formic acid) and solvent B (acetonitrile) in gradient elution at a flow rate of 0.3 mL/min with 40°C column oven temperature. Mobile phase solvent ‘A’ was prepared daily by weighing 0.12612 g of ammonium formate into 1000 mL volumetric flask, dissolved with 500 mL of purified water followed by 1 mL of formic acid then diluted to volume with purified water. The final solution was filtered through 0.45 µm membrane filter then degassed.
All chromatographic separations were performed using an agilent Zorbax RRHD Eclipse plus C18 column (1.8 µm, 2.1 mm × 100 mm, part #959758-902). Chromatographic separation was achieved in 8 minutes gradient elution. The initial mobile phase composition was 70% B, increasing to 90% B over 4 min, then held constant at 90% B for 2 min, and finally brought back to initial condition of 70% B in another 2 min followed by 1 minute re-equilibration. The injection volume for all samples was 20 µL.
Standard and quality control samples
Stock solutions (0.1 mg/mL) of IVM, IS and ALB (0.025 mg/mL) were prepared in acetonitrile. Stock solutions were diluted with acetonitrile to make working standard solutions which were further diluted to prepare the calibration standards and Quality Control (QC) samples. Matrix matched calibration standards were prepared by spiking 50 μL (100 ng/mL) from 1000 ng/mL IS and IVM intermediate solutions into 500 µL of human plasma and QC standards to obtain concentration levels of 0 ng/mL, 0.5 ng/mL, 1.0 ng/mL, 5.0 ng/mL, 10.0 ng/mL, 50.0 ng/mL, 100.0 ng/mL and 200 ng/mL. QC samples at four different concentrations; 0.5 ng/mL, 1.0 ng/mL, 80 ng/mL and 150 ng/mL of IVM which are hereby referred to as Lower Limit of Quantification (LLOQ), Low Quality Control (LQC), Middle Quality Control (MQC) and High Quality Control (HQC), respectively were prepared separately in sextuplicate. The stock, spiking calibration and QC solutions were kept at -30°C.
Sample preparation
IS (50 μL) and analytes were added into 15 mL conical polypropylene tubes containing 500 μL of human plasma and briefly vortexed to obtain the respective concentrations. To this mixture, 2 mL acetonitrile and 2 mL n-hexane were added, vortexed for 1 minute, sonicated for 6 minutes, vortexed again for 1 minute, incubated at -30°C for 10 minutes and centrifuged for 10 minutes at 4200xg × g at -2°C. Then, 2 mL of of the lower phase (acetonitrile) was transferred into another 15 ml conical bottomed polypropylene tube. The extracted sample solution was evaporated to dryness under a gentle stream of nitrogen at 70°C (for about 40 minutes), and the residue reconstituted with 0.5 mL of mobile phase at 1:1 ratio (A:B). The mixture was then vortexed for 1 minute, placed into an ultrasonic bath for 6 minutes, vortexed again for 1 minute and centrifuged at 4200xg × g at -2°C for 10 minutes. The supernatant was filtered through a 0.22 μm Polytetrafloroethylene (PTFE) syringe filter into LC vial and 20 μL was injected into LC system. The injection volume (20 μL) was within 10% of the column volume.
Method validation
The developed method was validated in terms of selectivity, specificity, linearity, low limit of quantification, accuracy, precision, recovery, matrix effect, carry over, dilution integrity and stability as described below. The acceptance criteria for validation parameters were based on the European Medicines Agency (EMA) and U.S Food and Drug Administration (FDA) bioanalytical method validation guidelines.
Selectivity and specificity
Selectivity and specificity were carried out by analyzing the six blank plasma samples spiked with IVM, ALB and IS. For specificity, six different lots of blank plasma were evaluated for any endogenous interference peaks at the retention times of the analytes and IS.
Linearity and carry over
The linearity of the method was evaluated using analyte spiked plasma samples in the concentration range of 0.5 ng/mL, 1.0 ng/mL, 5.0 ng/mL, 10.0 ng/mL, 50.0 ng/mL, 100.0 ng/mL and 200 ng/mL. Calibration curves were prepared from six different blank plasma samples, each obtained from different individuals. The results were fitted using linear regression analysis with theweighting factor (1/x²). The lowest concentration with RSD<20% was taken as LLOQ. Carry over was assessed by injecting three (3) blank human plasma and zero samples subsequently after injecting high concentration (200 ng/ml) sample of IVM and no significant peak (<20%) of the LLOQ) was observed.
Accuracy and precision
Within run and between run (intra and inter day) accuracy and precision were evaluated for three (3) consecutive days using six (6) replicates of QC (LLOQ (0.5 ng/mL), LQC (1 ng/mL), MQC (80.0 ng/mL) and HQC (150.0 ng/mL)) prepared on the same day.
Recovery
The recovery of analytes was calculated from six (6) replicates of spiked human plasma samples at each LQC (1ng/ml), MQC (80 ng/ml) and HQC (150 ng/ml) concentrations in extracted samples with pure authentic standards for IVM in reconstituted solvent.
Matrix effect
The matrix effect of human plasma constituents over the ionization of IVM and IS was determined by comparing the responses of the extracted plasma standard QC samples LQC (1 ng/ml), MQC (80 ng/ml) and HQC (150ng) in sextuplicate with the response of analytes from neat standard samples at equivalent concentrations. The Matrix Factor (MF) was calculated by determining the ratio of the peak area of the non-zero samples, to the corresponding peak area of the IVM and IS in selected diluents. The MF was calculated using the formula below;
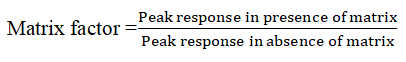
Dilution integrity
Dilution integrity was investigated to ensure that samples could be diluted with blank plasma without affecting the results. IVM and IS were spiked in human plasma at two concentration levels (300 ng/ml and 400 ng/ml) above the upper concentration level (200 ng/ml) and then diluted with pooled blank human plasma to obtain a minimum of quintuplicate determinants at each selected dilution factors of 2 and 5.
Stability
The stability of IVM in plasma samples were evaluated in triplicates at bench top storage (20°C for 8 h), following three freeze thaw cycles of the same (room temperature to -80°C to room temperature) and long term sample storage (-80°C for 30 days) at LQC (1.0 ng/ml), MQC (80.0 ng/ml) and HQC (150.0 ng/ml).
Human plasma sample analysis
The developed LC-MS/MS method was tested for its applicability by using human plasma samples obtained from individuals who received MDA drugs for the control of LF in endemic communities of Mkinga district, Tanga region, Tanzania. Study participants received a standard dose of IVM (Merck Sharpe and Dohme, Haarlem, Netherlands) as a single dose with ALB (400 mg). Two mL of venous blood were collected at 0, 2, 4 and 6 hours after drug administration from the antecubital arm vein. Blood was withdrawn using an indwelling intravenous catheter and collected into EDTA vacutainer tubes. The collected blood samples were centrifuged at 1000 rpm for 10 min to obtain plasma that were stored at -80°C until analysis. The involvment of human subject was approved by the Medical Research Coordinating Committee (MRCC) of the Tanzania National Institute for Medical Research (NIMR/HQ/R.8a/Vol. IX/2890).
Results and Discussion
Optimization of the LC-MS-MS method
The need for a functional and sensitive method for quantification of IVM is crucial in our setting since it is a part of MDA programme for the control of LF. This need is even compelled due to the fact that IVM is currently explored for the control of malaria which is a co-endemic infection in our setting. The method we outline here was developed based on the optimization of conditions used in previous methods and has proved to work well in our laboratory. The same has fullfilled the demands for IVM PK and PD studies planned in our settings. The MS/MS method development involved optimization of MRM conditions, including selection of precursor, product ions and optimization of Collision Energies (CE) as illustrated in Figure 2 below.

Figure 2: Chromatograms indicating the optimization of retention times, multiple reaction monitoring iron (without matrix) and mass spectra of the protonated molecules in positive electron spray ionization (ESI+) mode [M+NH4]+ for (A) Ivermectin; (B) Abamectin (IS) and; (C) Albendazole.
The internal standard (abamectin) used in this method was well separated from IVM and ALB. Figure 3 shows the selected reaction monitoring chromatograms for IVM, ALB and IS under optimized conditions.

Figure 3: Chromatograms from Multiple Reaction Monitoring ion (MRM). (A) Blank human plasma; (B) Plasma spiked with analytes at LLOQ; (C) Plasma spiked with analytes at HQC; (D) Superimposed chromatograms of blank human plasma, IVM, IS and ALB at LLOQ and HQC concentrations; (E) Plasma from individuals treated with IVM and ALB (2 hrs.) spiked with IS.
Several chromatographic conditions including different mobile phases, column and additives were tested for high sensitivity, improved peak resolution and shorter chromatographic times for IVM and IS. Optimum separation was achieved using mobile phase consisting of solvent A (2 mM ammonium formate with 0.1% formic acid) and solvent B (acetonitrile) in gradient elution at total flow rate of 0.3 mL/min with 40°C column oven temperature. The retention time for IVM was 6.2 min shorter than 7.5 min reported in the previous methods. The total chromatographic run time was 8 min which was less than 15 min reported in alternative method used to analyse dog plasma but comparable to a previous method by agilent in milk which was 6 minutes.
In the present method, sample extraction process was improved by introducing an incubation of diluted plasma sample at -30°C for 10 min. The incubation procedure enhances precipitation of proteins and fats which if left could be coextracted along with the analyte. Similarly the high centrifugation speed (4200 g) was able to remove some residual matrix through precipitation. Both incubation at low temperatiure and high centrifugation speed improved recovery and protected the column from being clogged up with residual matrix. The overall mean recovery and its respective RSD was 62.3% which is comperable to that reported in the previous methods but consistent between subjects (CV:<10.7%) confirming the usefulness of the extraction procedure (Table 2). In addition, the use of acetonitrile in the mobile phase improved stability and peak resolution as indicated in Figure 2.
| Concentration | % recovery | Accuracy (% bias) |
|---|---|---|
| LQC (1.0 ng/mL) | 62.5 | 10.7 |
| MQC (80 ng/mL) | 65.1 | 7.9 |
| HQC (150 ng/mL) | 59.2 | 5.3 |
Table 2: Recovery of IVM in human plasma.
Validation of the Method
The developed method was validated by evaluating selectivity, specificity, linearity, LLOQ, accuracy, precision, recovery, matrix effect, carry over, dilution integrity, stability of analytes and sample test solutions. Coefficients of variation and relative errors of less than 15% were considered acceptable, except for quantification limit where less than 20% was used as acceptance criteria as recommended elsewhere [15].
The specificity and selectivity of the method was evaluated by analyzing blank human plasma samples from six different sources in order to investigate potential interferences at the retention time of all three analytes. No coeluting peaks of >20% for LLOQ or >5% of the area of IS were observed. The retention times for ALB, IS and IVM, were 1.0, 4.3 and 6.2 min, respectively. This signifies that, our method is capable of detecting, differentiating and measuring IVM and ALB in the presence of potential interfering substances in the plasma matrix.
The linearity of the method extended from 0.5 ng/mL to 200 ng/mL, with correlation coefficient of higher than 0.999 for all validation runs (Figure 4). This range (0.5 ng/mL to 200 ng/mL) is sufficient to analyze IVM PK studies as reveled in the PK study (manuscript in drafting).

Figure 4: Calibration curve of IVM in (A) diluent and; (B) matrix-human plasma.
Above all, the RSDs for all concentrations including LLOQ were below 8%. Blank samples injected after the high standard (200 ng/ml), denoted insignificant peaks of IVM and IS in blank samples. The carry over response for IVM was below 5% of LLOQ and that of the IS was again below 1% demonstrating no significant carry over effect.
The developed method is accurate and precise as indicated by the bias (<14%) and RSD (<8%) for all analyzed samples as they were within the acceptable range (Table 3). This implies that, the closeness of the measured value and repeated individual measurements of the analyte obtained by the developed method were satisfactory.
| Level | Nominal conc. (ng/mL) | Within run (day 1) | Between run (day 2) | Between run (day 3) | Overall mean | ||||
|---|---|---|---|---|---|---|---|---|---|
| Precision (% RSD) | Accuracy (% Bias) | Precision (% RSD) | Accuracy (% Bias) | Precision (% RSD) | Accuracy (% Bias) | Precision (% RSD) | Accuracy (% Bias) | ||
| LLOQ | 0.5 | 4.2 | 7.0 | 5.2 | 16.1 | 14.8 | 18.2 | 8.1 | 13.8 |
| LQC | 1.0 | 4.8 | -0.9 | 2.4 | -1.2 | 8.3 | 12.7 | 5.2 | 3.5 |
| MQC | 80.0 | 4.5 | -2.2 | 7.9 | 8.7 | 11.2 | 11.9 | 7.9 | 6.1 |
| HQC | 150.0 | 2.3 | -7.2 | 5.5 | 6.6 | 7.6 | 2.5 | 5.1 | 0.6 |
Table 3: Investigation of within and between run precision (% RSD) and accuracy (% Bias) of IVM in human plasma.
There was no significant matrix effect observed as denoted by the matrix effect test for both analytes at LQC, MQC and HQC. Concentration levels in plasma were within ± 15% of the acceptable range (90.3%-107.8%) (Table 4). The precision for dilution integrity of 1:2 and 1:5 dilutions was within the acceptance limits of ± 15% for precision of coefficient of variation and 85.0%-115.0% for accuracy.
| Concentration | Av. peak area ratio | % RSD |
|---|---|---|
| LQC (1.0 ng/mL) | 0.0801 | 8.7 |
| MQC (80 ng/mL) | 3.0765 | 3.7 |
| HQC (150 ng/mL) | 5.5320 | 4.2 |
Table 4: Investigation of matrix effect.
IVM was stable in human plasma as signalled by the coefficient of variation which was within ± 15% of the actual concentration at all QC concentrations (Tables 5 and 6).
| Analyte | % Stability recoveries (mean ± SD) | ||
|---|---|---|---|
| Bench top (20°C, 8 h) | Freeze thaw (-80 ± 5°C, after 3 cycle) | Long term (-80 ± 5°C, 30 day) | |
| LQC | 108.1 ± 0.5 | 102.8 ± 0.3 | 103.7 ± 0.6 |
| MQC | 99.2 ± 1.0 | 101.0 ± 1.5 | 101.4 ± 2.6 |
| HQC | 100.4 ± 2.1 | 101.2 ± 2.3 | 101.8 ± 4.8 |
| Note: LQC: Lower Quality Control; MQC: Middle Quality Control; HQC: High Quality Control; SD: Standard Deviation. | |||
Table 5: Mean stability recoveries of ivermectin at different storage conditions in human plasma.
| Analyte | % Stability recoveries (mean ± SD) | ||
|---|---|---|---|
| Bench top (20°C, 8 h) | Freeze thaw (-80 ± 5°C, after 3 cycle) | Long term (-80 ± 5°C, 30 day) | |
| LQC | 102.1 ± 2.5 | 99.8 ± 4.7 | 96.8 ± 9.0 |
| MQC | 103.3 ± 2.8 | 102.9 ± 2.7 | 104.7 ± 4.1 |
| HQC | 103.2 ± 2.7 | 100.5 ± 0.6 | 102.1 ± 2.1 |
Table 6: Mean stability recoveries of abamectin at different storage conditions in human plasma.
Application of the developed method on field human plasma samples
The applicability of the method was tested using human plasma samples collected from the field among black individuals who were exposed to MDA in north-eastern part of Tanzania. A total of 468 individuals had their samples collected for PK study where 279 were males and 189 females with a median age of 33 (≥ 5) years. The median with Interquatile Range (IQR) for height, body weight and BMI of the individuals were 160 (151-1689) cm, 55 (43.5-65) kg and 20.43 (17.6-24.2) kg/m², respectively (Figure 5). The method worked efficiently and was able to generate reliable PK parameters. PK parameters were calculated based on a compartmental analysis using a modeling approach with NONMEM. Maximum IVM concentrations (54.25ng/mL), clearance (7.67 L/h), elimination half-life (55.25 hr) and time to reach maximun concentration (4 h) were consistent with the PK data reported in the previous literature. Figure 5 shows a concentration time curve for selected participants (n=10). Generally, the developed method is suitable for evaluating PK and PD disposition of IVM. We further used this method to assess pharmacokinetics of IVM that was given during MDA in one district in Tanzania (manuscript under development).

Figure 5: Concentration time curve of IVM obtained from plasma samples of 10 individuals who took part in MDA in north-eastern part of Tanzania (dots present mean plasma concentration and whiskers indicate 95% confidence interval of the mean).
Conclusion
The developed method applied positive ESI LC-MS/MS technique for high throughput quantification of IVM and simultaneous detection of ALB in human plasma. The method was highly precise, accurate, sensitive, selective and worked well in our laboratory. This method is a valuable tool for quantification of IVM and concomitant detection of ALB in human plasma samples which is critical for PK studies involving MDA drugs.
Acknowledgements
This study was conducted as part of the pharmacovigilance infrastructure and post marketing surveillance system capacity building for regional medicine regulatory harmonization in East Africa (PROFORMA) project funded by the European and Developing Countries Clinical Trials Partnership (EDCTP) 2 program, supported by the European Union (Grant number CSA2016S-1618) and the Swedish International Development Cooperation Agency (SIDA). We thank the funders for the support accorded to us.
References
- Shoop WL, et al. Vet Parasitol. 1995;59(2):139-156.
[Crossref] [Google Scholar] [PubMed]
- World Health Organization. WHO model list of essential medicines, 21st list. 2019.
- Kitzman D, et al. J Pharm Biomed Anal. 2006;40(4):1013-1020.
[Crossref] [Google Scholar] [PubMed]
- Conterno LO, et al. Cochrane Database Syst Rev. 2020;2020(4).
[Crossref]
- Sanbonsuge A, et al. Anal Methods. 2011;3(9):2160.
[Crossref]
- Liu Y, et al. Chin J Chromatogr. 2017;35(12):1276-1285.
[Crossref] [Google Scholar] [PubMed]
- Raich-Montiu J, et al. J Chromatogr A. 2008;1187(1-2):275-280.
[Crossref] [Google Scholar] [PubMed]
- Moschou IC, et al. J Chromatogr B Analyt Technol Biomed Life Sci. 2019;1104:134-140.
[Crossref] [Google Scholar] [PubMed]
- Morbidelli E, et al. MethodsX. 2018;5:1503-1507.
[Crossref] [Google Scholar] [PubMed]
- Durden DA. J Chromatogr B. 2007;850(1-2):134-146.
[Crossref] [Google Scholar] [PubMed]
- Inoue K, et al. J Sep Sci. 2009;32(21):3596-3602.
[Crossref] [Google Scholar] [PubMed]
- Chhonker YS, et al. Bioanalysis. 2018;10(22):1841-1852.
[Crossref] [Google Scholar] [PubMed]
- Duthaler U, et al. J Pharm Biomed Anal. 2019;172:18-25.
[Crossref] [Google Scholar] [PubMed]
- Smith G. Bioanalysis. 2012;4(8):865-868.
[Crossref] [Google Scholar] [PubMed]
- Duthaler U, et al. Br J Clin Pharmacol. 2019;85(3):626-633.
[Crossref]